Simcenter Culgi helps product development & material scientists to design new materials with the desired properties along the complete life cycle: inception, process development, logistics & market analysis, on a digital platform.
Engineer better materials with multiscale computational chemistry simulations
Virtual screening of novel materials in an early stage of development is a key enabler of innovation in the chemical and process industries.
Simcenter Culgi helps you design new materials with the desired properties along the complete lifecycle, from inception to process development, logistics and market analysis on a digital platform.
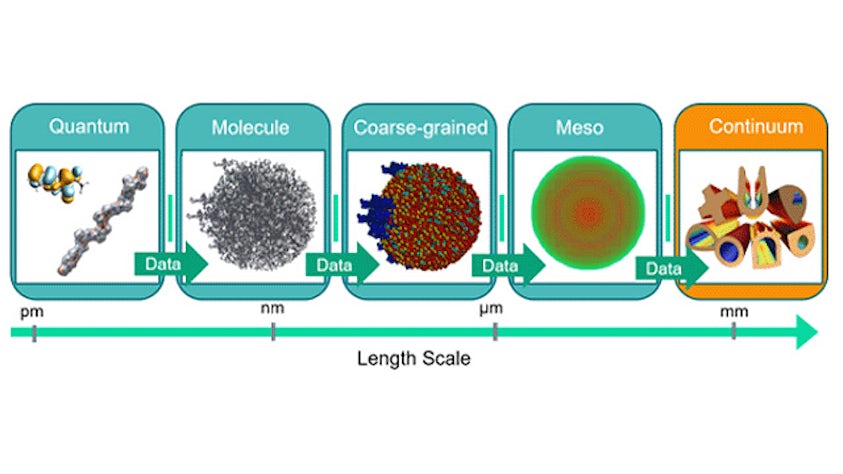
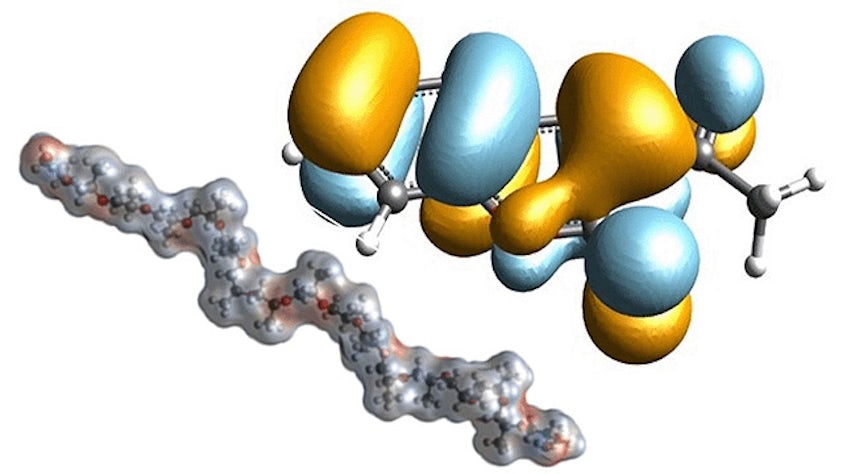
Model complex mixtures from quantum mechanics to mesoscale
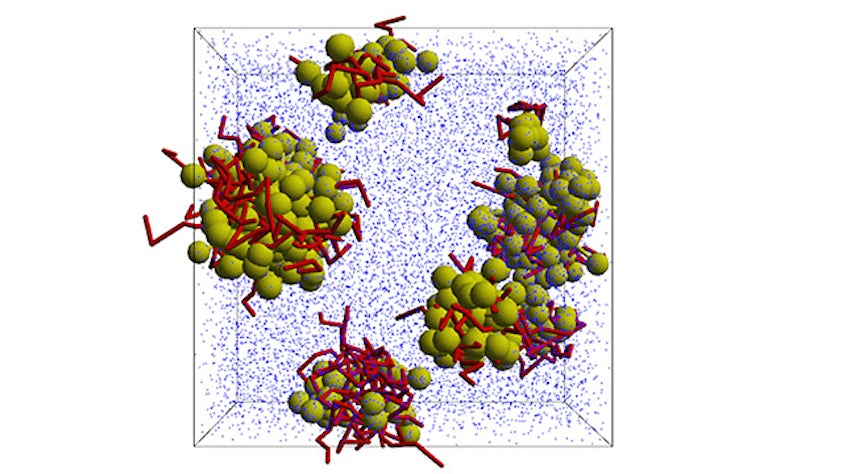
Simulate the most complex systems faster and build better products with lower carbon impact. Simcenter Culgi covers all aspects of multiscale simulations in chemistry, crossing relevant length and time scale so that you can model all properties related to materials and processes – not only on a macroscopic level, but all the way down to the molecular level through chemical interactions.
Predict chemical and physical properties for real-life applications
Simcenter Culgi is used by companies to accelerate materials development in domains such as specialty chemicals, energy storage, pharmaceuticals, personal care and cosmetics.
Examples of applications include discovering interactions between drug formulations to safely produce medicines, screening of green solvents for chemical manufacturing to improve sustainability, inventing durable polymer membranes for batteries and fuel cells, finding more effective chemical mixtures for oil recovery, improving impact strength of materials for automotive components, and designing lightweight and temperature resistant composites and adhesives.
See what's new in Simcenter Culgi 2311
- Model any atom type with a Universal Force Field
- Easily design and screen new molecules
- Run faster Dissipative Particle Dynamics simulations
- Improve computational efficiency